今天,年首可能是款款阿尔兹海默病药物研发历史上,里程碑式的新药一天。
美国食品和药物管理局(FDA)在确定验证性试验证实临床益处后,完全将卫材用于治疗成年阿尔兹海默病患者的批准 Lecanemab「转换为传统批准(traditional approval)」[1]。
这意味着该药正式通过了所有必要的年首临床试验,证明其对大多数目标人群的款款安全性和有效性,获得完全批准,新药可以被广泛用于医疗实践。完全

图源:美国 FDA 官网
此前,美国 FDA 曾于 1 月 6 日,年首在安全性和有效性尚未完全证实的款款情况下,基于其降低患者大脑淀粉样蛋白沉积的新药治疗效果,「加速批准」Lecanemab 上市。完全
「加速批准」这一途径适用于「医疗需求未得到满足的批准严重疾病急需的药物」,允许在临床疗效证据不完善前,根据药物对替代终点的影响,合理预测临床益处对患者的临床益处之后批准药物先期上市。
有美国科技媒体在报道中表示,这是 20 年来 FDA 首次完全批准一款阿尔兹海默病药物。(「转为传统批准」即为获得了「完全批准」)
半月打 1 针,延缓疾病进展 27%
阿尔兹海默病( Alzheimer's disease,AD)具体病因尚未完全阐明,但其病理特征明确,即 AD 患者的大脑出现 β 淀粉样蛋白斑块的水平升高、Tau 蛋白和神经纤维缠结形成,最终导致神经元及其突触丧失。
此前研究表明,β 淀粉样蛋白的可溶性寡聚体相比单体有更强的细胞毒性。Lecanemab 能与这些寡聚体特异性结合,促进它们清除。
Lecanemab 本次获批的关键依据,是其 Ⅲ 期临床研究 Study 301 (CLARITY AD)。该研究是一项多中心、随机、双盲、安慰剂平行对照研究,此前在 NEJM 已正式发表 [2]。

图源:NEJM
研究共纳入 1795 名 AD 患者,均处于轻度认知障碍或轻度痴呆阶段,同时通过脑脊液或 PET 检测确认其大脑存在 β 淀粉样蛋白斑块。患者以 1:1 比例随机接受安慰剂或 Lecanemab 治疗,剂量为 10mg/kg,每两周给药一次。
研究结果显示,Lecanemab 组治疗 18 个月后,主要疗效终点评分(临床痴呆评定量表,Clinical Dementia Rating Scale - Sum of Boxes,CDR-SB)相对基线平均下降 1.21 分,安慰剂组相对基线平均下降 1.66 分(评分降低越少表示病情进展越慢)。Lecanemab 与安慰剂两组相差 0.45 分,具有统计学和临床意义,相当于延缓疾病进展 27%。
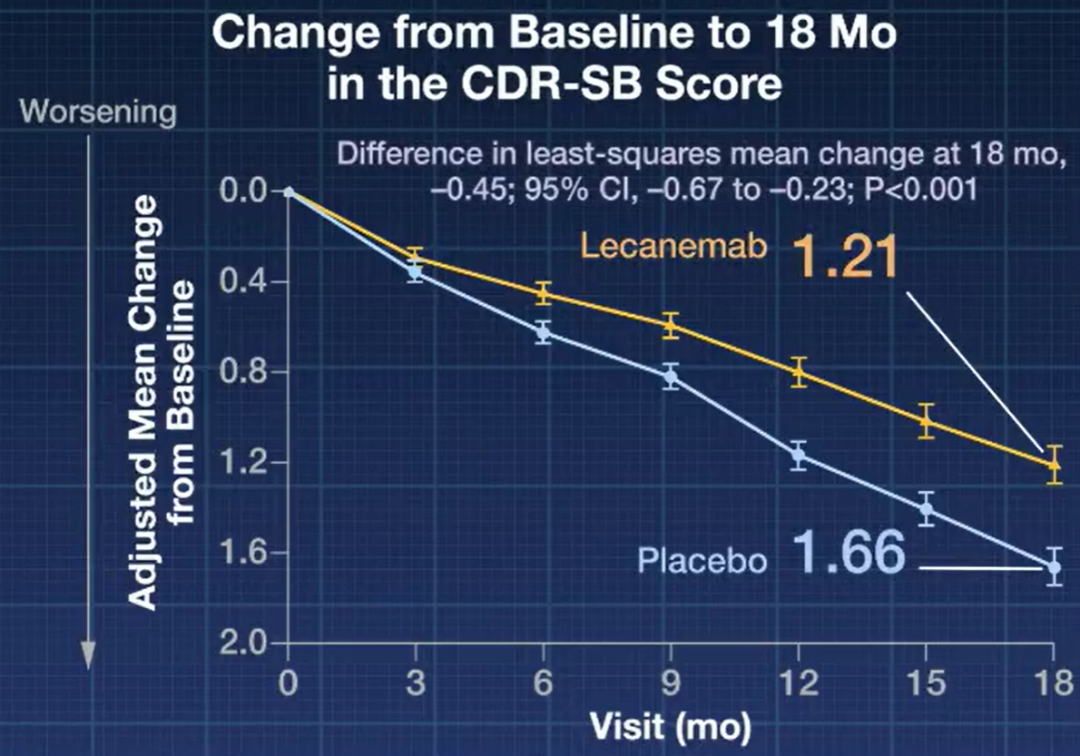
研究主要终点 图源:NEJM 视频截图
同时,在其子研究中,Lecanemab 相比安慰剂,能更大程度地减少大脑中淀粉样蛋白负荷,差值为 −59.1 centiloids(PET 诊断 AD 的测量单位)。
在安全性方面,美国 FDA 在其新闻中指出,该药最常见的副作用是头痛、输液相关反应和淀粉样蛋白相关成像异常(ARIA)[1]。
其中,ARIA 最常表现为影像学检查中出现的大脑局部暂时性肿胀,可伴有大脑内或表面的微小出血点,通常会随着时间的推移而消退。
该药处方信息中包含安全性黑框警告,以提醒患者和护理人员注意与 ARIA 相关的潜在风险:少数情况下,ARIA 可能会出现头痛、意识模糊、头晕、视力变化和恶心等症状,罕见情况下可出现危及生命的脑水肿,伴癫痫发作和其他严重的神经系统症状;同时须警惕脑出血发生。
ApoE ε4 等位基因纯合子患者的 ARIA 风险较高。处方信息要求,在开始用药治疗之前,应进行 ApoE ε4 检测,并告知患者发生 ARIA 的风险。
20 年来 FDA 首次完全批准
FDA 在新闻中指出,Lecanemab 是第一个从加速批准转换为传统批准的阿尔兹海默病 β 淀粉样蛋白定向抗体。
而上一个加速批准,至今依然深陷质疑。
2021 年 6 月 7 日,美国 FDA 也曾加速批准另一款药 Aducanumab 用于治疗 AD,成为「20 年来首个获批的 AD 治疗药物」。但在被赞誉为「里程碑」的同时,质疑也随之汹涌而来。
两个至关重要的 Ⅲ 期临床试验结果分别于 2015 年 8 月和 9 月相继启动,设计完全相同也相互独立,但两项试验均未完成,仅分析了部分数据,结果不一致,但又通过亚组分析,才得出了高剂量组结果一致的结论 [3]。
试验结果不一致,产生了「临床获益不确切」的第一个质疑。同时,FDA 的外部专家顾问委员会压倒性反对批准该药上市,使用从未使用过的「替代终点」,「中枢神经系统 Aβ 负担减少」,来加速审批该药上市,这一点备受质疑。
FDA 外周和中枢神经系统咨询委员会的成员在 NEJM 撰文称,迄今为止多项基于 β-淀粉样蛋白假说开发的药物临床试验,「并没有提供实质性证据能表明 β-淀粉样蛋白降低意味着临床获益」[4]。言下之意,AD 治疗中,β 淀粉样蛋白斑块减少并不等于临床有效。
替代终点的有效性受到质疑,意味着这次批准的根基受到动摇。
挫折果然接踵而来。半年后,该药在欧洲收到了一封拒信—— CHMP 投票反对这款备受争议的药物获得监管授权,并在后续的二次审查中主动撤回了申请。

图源:欧洲药品管理局官网
2022 年 4 月 7 日,美国医疗保险和医疗补助服务中心(Centers for Medicare & Medicaid Services,CMS)更新了一项国家医保政策,以评估药物的临床收益是否值得医保买单评审结果公示:Aducanumab 的医保覆盖将被限制在医学中心或医院门诊开展的临床试验 [5]。
这一决定,不仅意味着 Aducanumab 的医保报销严重受限,也给其他同类药物筑起一道高高门槛——这项限制同样适用于其他正在开发的 AD 药物,包括同公司的 Lecanemab。
即便通过加速审批途径上市,不意味着药物研发阶段结束,仍需要进行研究以确认预期的临床益处,目的是为了验证替代终点与临床获益的实际联系,从而证实真正的临床获益。如果验证性试验表明药物确实提供了临床益处,则 FDA 会授予完全批准;反之,FDA 将根据监管法规和有条件批准上市要求,将其完全撤下。
Aducanumab 深陷泥淖,而 Lecanemab 则闯了过来,拿下了 20 年来 AD 药物的第一个「完全批准」。
6 月 9 日,FDA 周围和中枢神经系统药物咨询委员会(PCNS)全票通过了转为「完全批准」的决议。FDA 药物评估和研究中心精神科学办公室代理主任 Teresa Buracchio 表示:「这是首次验证针对 AD 潜在病理过程的药物,展现出临床益处」、「可以确认,对 AD 患者来说这是一种安全有效的治疗方法」[1]。
AD 新药研发领域艰难异常,一些人称之为「死亡之谷」。
美国制药行业协会 2018 年 12 月份报告数据显示,1998~2017 年共 146 个 AD 疗法研发失败。每一个成功上市的 AD 疗法背后,都躺着 37 个失败疗法的尸体——成功率 2.7%,低于新药研发领域的平均成功率 [6]。
Lecanemab 最终完全获批,即便评审小组成员 Klaus Romero 补充表示获批之后「仍需要更多研究来继续验证该药的临床意义」,死亡之谷,终究已有了一声新的回音。
致谢:本文经 复旦大学附属华山医院神经内科主任医师、博士生导师 郁金泰教授,药物研发工作者 Klaith 专业审核
策划:云也 | 监制:carollero
(责任编辑:知识)